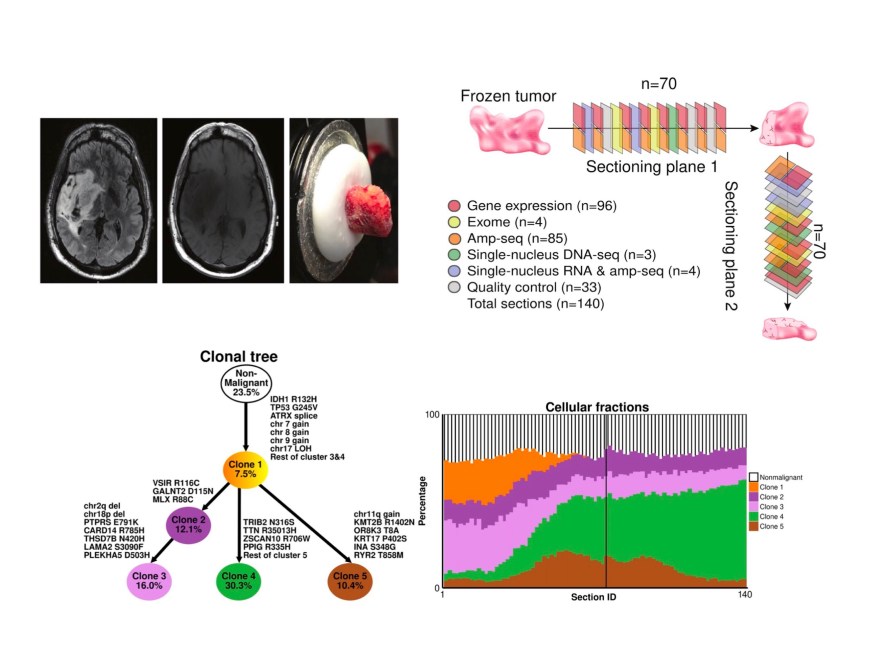
Full reference: Cancers (Basel). 2024 Jul 1;16(13):2429. doi: 10.3390/cancers16132429.PMID: 39001492
Cancerous tumors may contain billions of cells including distinct malignant clones and nonmalignant cell types. Clarifying the evolutionary histories, prevalence, and defining molecular features of these cells is essential for improving clinical outcomes, since intratumoral heterogeneity provides fuel for acquired resistance to targeted therapies. Here we present a statistically motivated strategy for deconstructing intratumoral heterogeneity through multiomic and multiscale analysis of serial tumor sections (MOMA). By combining deep sampling of IDH-mutant astrocytomas with integrative analysis of single-nucleotide variants, copy-number variants, and gene expression, we reconstruct and validate the phylogenies, spatial distributions, and transcriptional profiles of distinct malignant clones, which are not observed in normal human brain samples. Importantly, by genotyping nuclei analyzed by single-nucleus RNA-seq for truncal mutations identified from bulk tumor sections, we show that commonly used algorithms for inferring malignancy from single-cell transcriptomes may be inaccurate. Furthermore, we demonstrate how correlating gene expression with tumor purity in bulk samples provides the same information as differential expression analysis of malignant versus nonmalignant cells and use this approach to identify a core set of genes that is consistently expressed by astrocytoma truncal clones, including AKR1C3, whose expression is associated with poor outcomes in several types of cancer. In summary, MOMA provides a robust and flexible strategy for precisely deconstructing intratumoral heterogeneity in clinical specimens and clarifying the molecular profiles of distinct cellular populations in any kind of solid tumor.
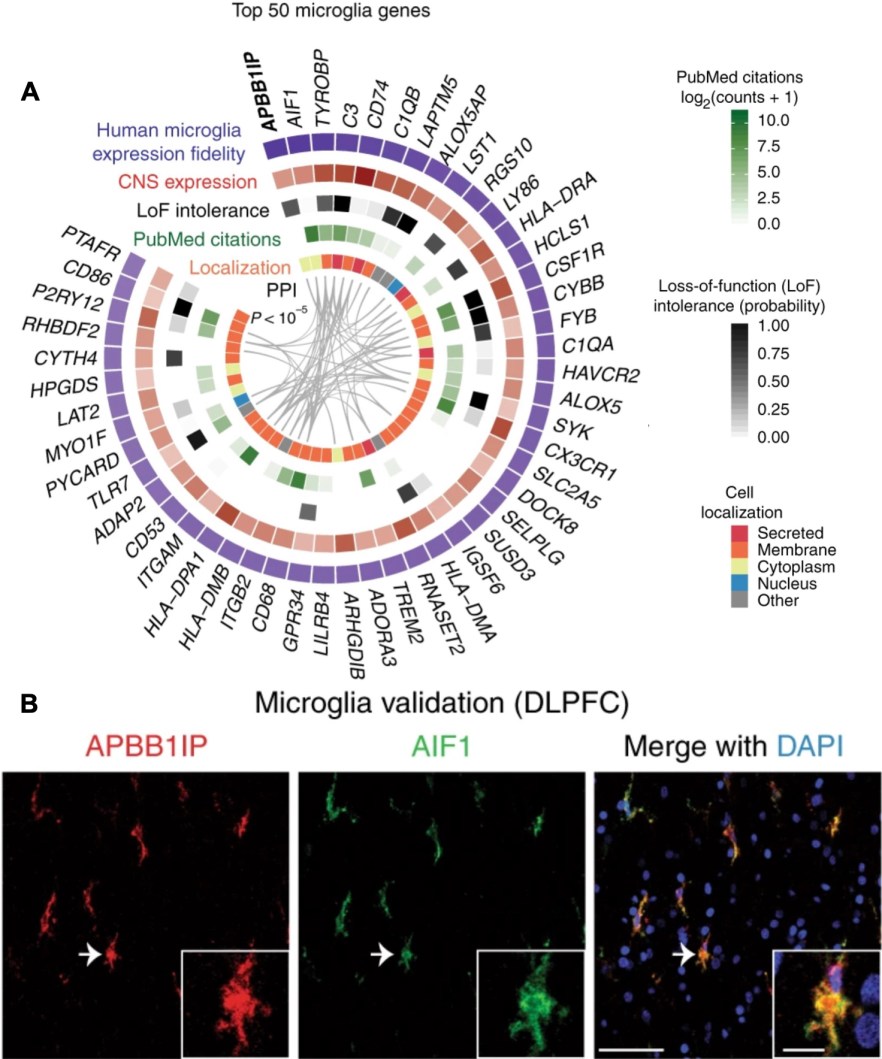
Full reference: Kelley et al. Variation among intact tissue samples reveals the core transcriptional identities of human CNS cell classes. Nature Neuroscience (2018) Sep;21(9):1171-1184. doi: 10.1038/s41593-018-0216-z
It is widely assumed that cells must be physically isolated to study their molecular profiles. However, intact tissue samples naturally exhibit variation in cellular composition, which drives covariation of cell-class-specific molecular features. By analyzing transcriptional covariation in 7,221 intact CNS samples from 840 neurotypical individuals, representing billions of cells, we reveal the core transcriptional identities of major CNS cell classes in humans. By modeling intact CNS transcriptomes as a function of variation in cellular composition, we identify cell-class-specific transcriptional differences in Alzheimer’s disease, among brain regions, and between species. Among these, we show that PMP2 is expressed by human but not mouse astrocytes and significantly increases mouse astrocyte size upon ectopic expression in vivo, causing them to more closely resemble their human counterparts. Our work is available as an online resource (http://oldhamlab.ctec.ucsf.edu/) and provides a generalizable strategy for determining the core molecular features of cellular identity in intact biological systems.
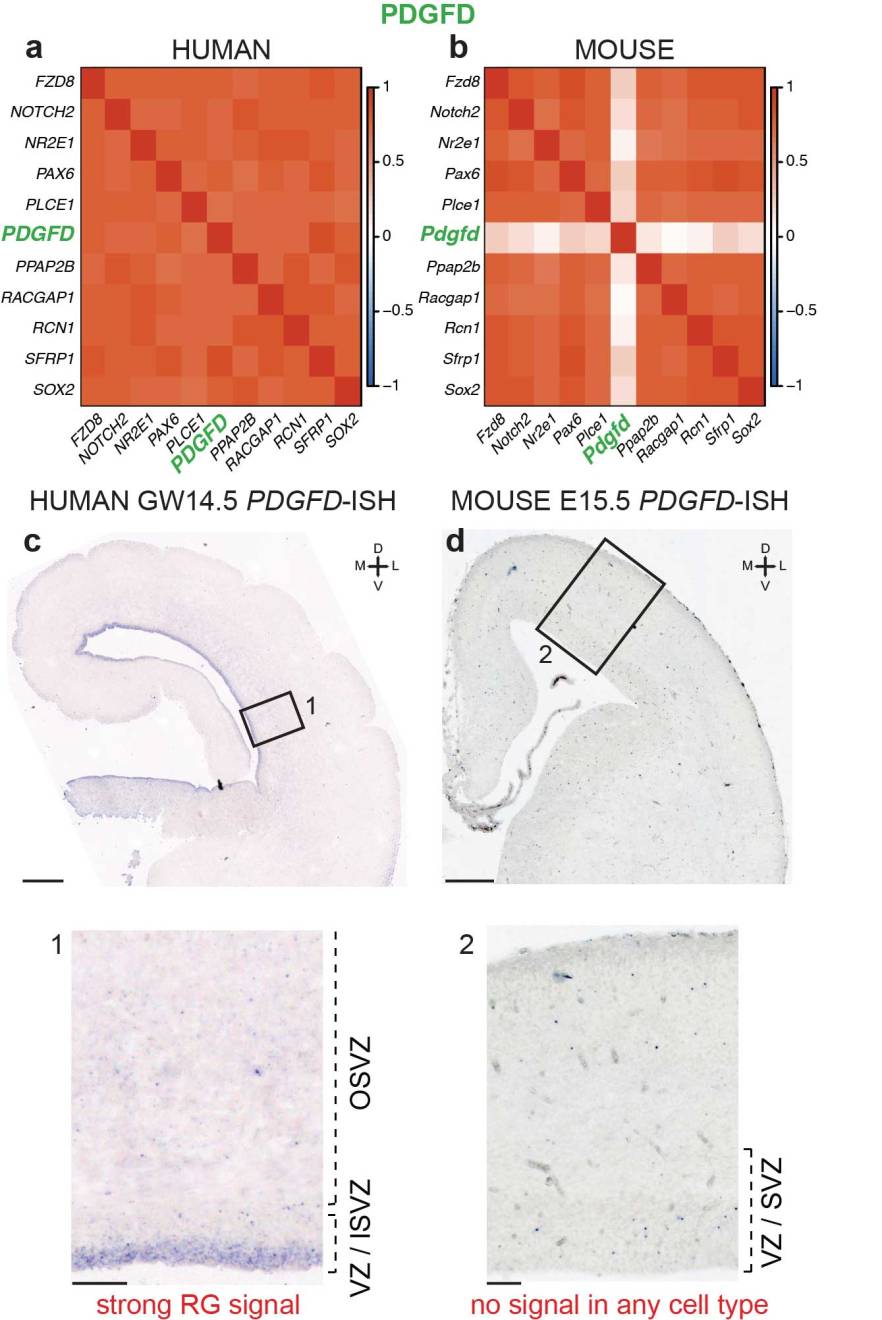
Radial glia require PDGFD–PDGFRb signalling in human but not mouse neocortex
Full reference: Lui et al. Radial glia require PDGFD-PDGFRß signaling in human but not mouse neocortex. Nature (2014) vol. 515 (7526) pp. 264-8
Evolutionary expansion of the human neocortex underlies many of our unique mental abilities. This expansion has been attributed to the increased proliferative potential of radial glia (RG; neural stem cells) and their subventricular dispersion from the periventricular niche during neocortical development. Such adaptations may have evolved through gene expression changes in RG. However, whether or how RG gene expression varies between humans and other species is unknown. Here we show that the transcriptional profiles of human and mouse neocortical RG are broadly conserved during neurogenesis, yet diverge for specific signalling pathways. By analysing differential gene co-expression relationships between the species, we demonstrate that the growth factor PDGFD is specifically expressed by RG in human, but not mouse, corticogenesis. We also show that the expression domain of PDGFRb, the cognate receptor for PDGFD, is evolutionarily divergent, with high expression in the germinal region of dorsal human neocortex but not in the mouse. Pharmacological inhibition of PDGFD–PDGFRb signalling in slice culture prevents normal cell cycle progression of neocortical RG in human, but not mouse. Conversely, injection of recombinant PDGFD or ectopic expression of constitutively active PDGFRb in developing mouse neocortex increases the proportion of RG and their subventricular dispersion. These findings highlight the requirement of PDGFD–PDGFRb sig- nalling for human neocortical development and suggest that local production of growth factors by RG supports the expanded germinal region and progenitor heterogeneity of species with large brains.